Cell culturing is one system that enables easy access to living neurons. Primary culture is one that is produced from tissue dissected from an organism. Different types of primary neuronal cultures can be made from a variety of animals, developmental stages and nervous system tissues. This protocol provides step-by-step instructions for dissecting and culturing hippocampal primary neurons in the mouse.
1. Dissection of Rat Hippocampi
●Take the pregnant mouse on the 18th day of embryonic period (E18) (the cell culture can be the newborn mouse with P0-P1, and anesthetize it on ice), and anesthetize with ether or isoflurane mixed gas.
Note: It is not recommended to anesthetize the mouse by intraperitoneal injection of chloral hydrate or pentobarbital sodium, it is because intraperitoneal injection produces a greater impact on embryonic mouse.
●Recover embryos via cesarean section quickly on ice. Cut off the head of the embryo and place it in a dissection medium on ice.
●Remove skin and skull and take out the brain connected to the cerebellum, then place it in another dish containing dissection medium. Keep the dish on ice.
●Divide the brain into two halves, and cut off at the base of the ventral side that connects with the cerebellum during the division, so that the hippocampus is easily exposed.
●Remove the meninges and cut out the hippocampus with small scissors. Because there are many cortical cells, you can remove the cortical tissue close to the cerebellum.
Note: Do not squeeze the brain tissue with tweezers, as this will cause a large area of cell death, which is not conducive to subsequent culture.
●Place the hippocampal tissue in a new dish on ice. Cut the tissue into pieces with small scissors in the dish.
2. Dissociation and Culture of Rat Hippocampal Neurons
●Transfer the tissue pieces with dissection medium to a 5 μl Eppendorf tube. Standing for a period of time until the tissue have sunk to the bottom,then discard the clear supernatant.
Note:No need to suck up completely.
●Mix trypsin, DNase and digestion solution at the volume ratio of 1: 2: 3 in another 5 μl Eppendorf tube, filter with a strainer and add it to the tissue pieces.
●Incubate for 7–8 minutes in a 37 °C cell culture incubator and mix well every 2 minutes.
Note: The digestion time depends on the concentration of the enzyme and should not be over 15 minutes.
●Prepare trypsin inhibitor (TI) and dissection medium mixture at the volume ratio of 1:4 (or 5% FBS+Neurobasal+B27) in another 5 μl Eppendorf tube. Add a certain amount of it to a pre-coated dish.
●Centrifuge (or natural sedimentation) the digested tissue pieces, then discard the remaining liquid on the upper layer. Add the mixture in the previous step to stop digestion at 37 ℃ for 2-3 minutes.
Note: the next steps are required to be completed on ice.
●Centrifuge (or natural sedimentation) until the tissue have sunk to the bottom, then discard the clear supernatant. Add DNase (which can be diluted with dissection medium if needed) and re-suspended the cell pellet.
●Centrifuge for 5-6 minutes and discard the supernatant. Add 2-3 ml dissection medium, and pipette up and down a few times to help with tissues dissociation.
●Count the number of cells after 10 times dilution by mixture of 10 μl suspension and 90 μl trypan blue.
●Plate the neurons on the prepared culture plates according to the desired seeding density, then mix it well.
Note: In general,the brain of a mouse typically yields 2 ×10
7-3×10
7 neurons,and neurons cultured for 3-9 days in vitro can be all infected by lentivirus.
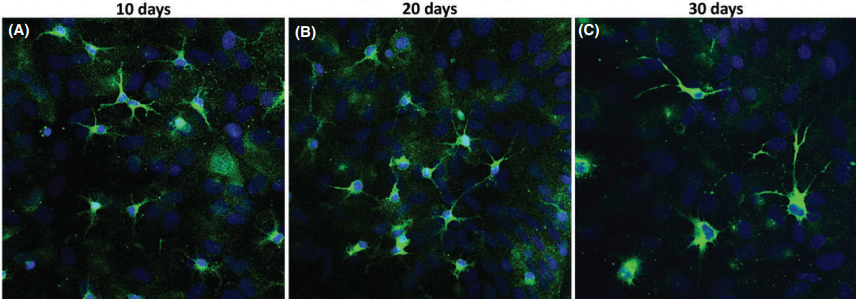 Hippocampal primary culture immunostained for MAP2 (green) and Hoechst (blue) after 10, 20, and 30 days(CNS Neuroscience & Therapeutics 22 (2016) 440–450)
Hippocampal primary culture immunostained for MAP2 (green) and Hoechst (blue) after 10, 20, and 30 days(CNS Neuroscience & Therapeutics 22 (2016) 440–450)
If you have any questions,please email us at
sales@brainvta.com.

 sales@brainvta.com
sales@brainvta.com