Cortical neural cell cultures are an important model system for studying neuronal development and function, neurotoxicity screening, drug discovery, and mechanisms of neurological diseases. Proper development and survival of neurons, requires specific growth factors, signaling molecules, peptides, and vitamins. This protocol provides step-by-step instructions for dissecting and culturing cortical neurons in mouse.
(1) Coating and washing culture plates
●Coat the culture plate with 0.1 mg/ml poly-D-lysine overnight in the cell culture incubator.
●Rinse three times with sterile tri-distilled water and dry it in a laminar flow bench for use.
●Sterilize general surgical instruments, filter paper and screen mesh with high pressure but not precision surgical instruments. Sterilize with ultraviolet irradiation for 30 minutes before use, and wipe with 75% ethanol after use.
(2) Dissection of mouse cortical tissue
●Decapitation: Newborn (<24h) mouse is sterilized with 75% ethanol. Decapitate each mouse at the head/neck junction under aseptic conditions, then pick up whole brain from the mouse and place it in 75% alcohol for about 1 min. (keep the operation on ice)
●Take out the brain: Remove skin and skull from anterior to posterior with micro-tweezers under a microscope, and finally peel back the brain with the forceps to keep it intact as possible.
●Take out the cortex: Cut off the brainstem and divide it into two, then take out the hippocampus. The upper part is the cortex, remove the meninges and transfer tissues to a dish containing 1xHBSS (Hank’s balanced salt solution). The tissues are cut into as small piece as possible with micro-scissors.
●Digestion: Transfer tissues to a 24-well plate, discard the dissecting solution and add 0.25% trypsin (containing 6 μL/mL DNase solution to avoid viscosity affecting the digestion effect), 50 μL/well, incubate in a 37 °C incubator for 30 minutes and shake well every 10 minutes during this period.
●Washing: Transfer the cortical tissue pieces into the centrifuge tube and discard the remaining digestion solution, then wash once with the inoculation solution and discard the supernatant.
●Pipetting: Re-suspend the cortical tissue pieces with 1 ml of inoculation solution. Slowly and gently pipette (1 ml imported pipette tip) the tissue pieces up and down 10 times. Standing for a period of time until the tissue have sunk to the bottom then, and transfer the supernatant to a centrifuge tube for use. Repeat 3 times for tissues dissociation and discard the remaining pieces.
●Screen: The supernatant is filtered through a 200-mesh strainer to obtain single cell suspension.
●Inoculation: dilute the filtrate by ten times and count the number of the cells. The inoculation density is about 400,000 per 12-well plate.
(3) Exchanging media in cortical neuron cultures
●Change the media: After culturing for 4-6 hours until the cells adhere to the wall. Wash the cell pellet with DMEM high-sugar medium, then replace it with Neuronal Base Media and culture for 3 days to observe the growth of neurons.
●Change the media: replace with cytarabine culture medium (final concentration 2.5 μg/mL, change half of the medium) for 3 days to inhibit the growth of glial cells and other cells, and obtain purely cultured primary neural cells.
●Change the media: Change the culture medium once every 3 days, changing half of the medium each time. Cultivate for 7-9 days for experiment.
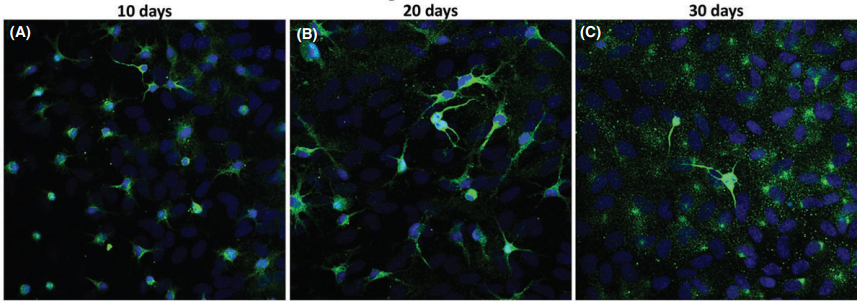 Cortical neurons cultures immunostained for MAP2 (green) and Hoechst (blue) after 10, 20, and 30 days(CNS Neuroscience & Therapeutics 22 (2016) 440–450)
Cortical neurons cultures immunostained for MAP2 (green) and Hoechst (blue) after 10, 20, and 30 days(CNS Neuroscience & Therapeutics 22 (2016) 440–450)
If you have any questions,please email us at
sales@brainvta.com.

 sales@brainvta.com
sales@brainvta.com